SLURM
Connections and Agents
Lots of stuff to compute and only a laptop at hand? Don’t worry, most probably your university has a compute cluster that can do most of the work for you. Maybe, you already figured out how to connect to the cluster via SSH. But how do we actually run a computation on a cluster? This is what this post is about.
In this post, we focus on scientific clusters that manage workload with SLURM.
TL;DR
- SLURM is a workload manager that makes sure that computations get the correct resources and that computation time is allocated fairly among users.
- Make sure jobs store data on the
scratchordatapartition of the cluster (if it has one) and don’t write GB of data into your home directory. Usually, home directories are backed up and only have limited amount of storage available. sbatchsubmits ansbatch-formatted bash script to a compute node (recommended way to submit jobs).srun <command>executes the followingcommandon a compute node of the cluster.squeuelists all currently running jobs
Structure of a cluster
Compute clusters are a bit bigger than the average home computer. The amount of main memory (RAM) of a cluster node can have the size of a normal hard drive (1TB of RAM is not unheard of). Let’s have a quick look at what a cluster looks under the hood.
Usually, you log into one or two special nodes of the cluster: the login nodes. These nodes are not meant for computations, but for management tasks like small tests, compiling, moving data, etc. They are also used to send jobs via the job management system, i.e. SLURM, to the compute nodes. These are usually a bit beefier and do the actual heavy lifting. Except for special cases, like debugging, there is usually no need to log into these compute nodes. Usually, login and compute nodes have access to network storage.
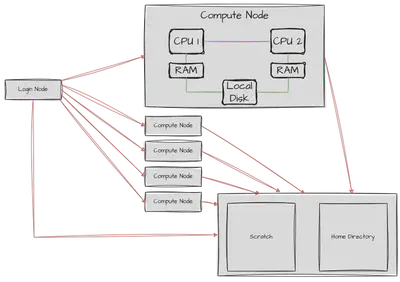
$HOME partitions and scratch partition.
When working on the cluster, your \$HOME partition is backed up automatically.
However, it has only limited capacity.
If you are submitting compute jobs, you should move them to the scratch partition of the cluster.
For xmaris, one of the physics clusters in Leiden, it is located at /marisdata/<username>.
For alice, the Leiden university cluster, it is at /data1/<username>.
When you use it the first time, you might have to create the folder with your username.
There is (to my knowledge) no quota on this partition and its capacity is in the TB regime.
However, it is not back-up.
So don’t store anything here that you can’t recalculate and move important data to $HOME.
Note, however, the home directory is rather small (approx. 10 GB)
For an example of the actual specifications of a cluster, have a look here for the xmaris compute cluster at the Lorentz Institute in Leiden or here for alice.
Running jobs
Most scientific clusters use SLURM as a job management system. Here, we focus on giving a practical introduction to using SLURM. If are looking for a more technical introduction, have a look here.
The idea of a job system is that not everyone can work on the same node and that different jobs should be scheduled in such a way that the cluster is used most efficiently. You use the login node to add jobs to the queue of jobs that will be executed on the compute nodes.
To use SLURM, we need a bit of vocabulary. Let’s go with a couple of hand-wavy definitions to get the idea across.
SLURM defines certain terms to manage the allocation of cluster resources. The biggest chunk that you can allocate are one or multiple nodes. A node has usually multiple CPUs each equipped with multiple processors. Thus, if you allocate multiple nodes, you will have to implement some way to communicate over network (usually MPI). If you didn’t do this, don’t allocate multiple nodes for one job.
Starting multiple tasks you have to be able to communicate between multiple processes on the same node. That is also usually done via MPI.
The last layer is allocating processors to a job. Here, a processor refers to a physical core, not a packaged CPU. The communication between multiple cores is usually managed by most libraries that you might be using (numpy, Lapack, BLAS, etc) via OpenMP or similar libraries. Thus, allocating multiple cores for a job makes a lot of sense.
Starting a job
Enough theory, let’s start a job.
The usual way to start a job is the sbatch command which is called with a script that details the configuration of the job as well as the program to be called
sbatch <slurmscript>
A SLURM script might look like this:
#!/bin/bash -l
# Standard output and error:
#SBATCH -o ./tjob.out.%j
#SBATCH -e ./tjob.err.%j
# Initial working directory:
#SBATCH -D ./
# Job Name:
#SBATCH -J NAME_FOR_YOUR_JOB # You might want to change this line
# Queue (Partition):
#SBATCH --partition=compIntel #This depends on your cluster, check with `sinfo`
# Number of nodes and MPI tasks per node:
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task=8
#
#SBATCH --mail-type=FAIL
#SBATCH --mail-user=YOUR_EMAIL # You might want to change this line
#
# Wall clock limit:
#SBATCH --time=00:02:00
#Load some modules
module load Python/3.9.5 #this depends on your cluster, check with `module av`
# Run the program:
echo "TEST" # You might want to change this line
If you are in a hurry, you can also start a job with
srun <command>
However, the setup here dominated by default values and you might get the wrong kind of node or the wrong time limits.
Most parameters that are provided to sbatch can be provided to srun via the command line.
For reproducability it is much nicer to use sbatch.
Observing the cluster workload
The command squeue tells you what jobs are currently running or pending on the cluster
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
1990265_15 ibIntel w_30_lj3 flor R 48:27 1 maris084
1990265_14 ibIntel w_30_lj3 flor R 48:29 1 maris085
1990265_13 ibIntel w_30_lj3 flor R 49:27 1 maris085
1990265_12 ibIntel w_30_lj3 flor R 49:40 1 maris085
1990265_11 ibIntel w_30_lj3 flor R 49:41 1 maris085
1990265_10 ibIntel w_30_lj3 flor R 50:06 1 maris085
1990265_9 ibIntel w_30_lj3 flor R 50:10 1 maris085
1990265_8 ibIntel w_30_lj3 flor R 50:26 1 maris085
1990265_7 ibIntel w_30_lj3 flor R 50:56 1 maris085
1990265_6 ibIntel w_30_lj3 flor R 51:25 1 maris084
1990265_5 ibIntel w_30_lj3 flor R 51:28 1 maris085
1990265_3 ibIntel w_30_lj3 flor R 51:36 1 maris085
In case, you want to restrict it to your user, use the -u <username> option.
An interesting alias might be:
alias squeuel='squeue --format="%.18i %.9P %.30j %.8u %.2t %.10M %.6D %R" -u <username>'
Cancel a job
In case you submitted a job and realize that you don’t want it executed or want to abort it, use
scancel <jobid>
You get the <jobid> from the output of the squeue command.
If you feel like cancelling all your jobs
scancel -u <username>
Other commands
If you are curious about the usage of SLURM partitions, you can have a look at the output of
sinfo
Further information about the number of CPUs used by your jobs and other details are printed by
sacct
An overview by the Lorentz institute of all relevant cluster commands can be found on this page.
A similar overview for the alice cluster is provided here.
Useful alias commands
alias sinfot='sinfo -M all -N -o "%25P %16N %16T %16C %10z %.10e/%10m %10g %b"'
This lists the most important information about the cluster in an easy-to read manner.alias squeuel='squeue --format="%.18i %.9P %.30j %.8u %.2t %.10M %.6D %R" -u <username>'After substituting<username>with your username, you will get a nice overview of your jobs only, with extra space for job names.
These alias commands could go into your .bashrc (assuming that your cluster uses bash as a default shell) to make working with SLURM a bit more comfortable.
Commands in this post
sbatch: Start a job described in a bash file (recommended)srun: Start a job directly from the command linesqueue: List the queue of jobs on the clustersqueue -u <username>: List the queue of jobs by user<username>sinfo: List information about the cluster, its partitions, etcsacct: Details about the history of jobs by a user
Patrick Emonts
Junior Research Group Leader
My research interests include tensor networks, lattice gauge theories and quantum information.